从敌人到朋友,动物毒素类上市及在研药物盘点
2017年09月14日 10:58:41
来源:品途网

原标题:从敌人到朋友,动物毒素类上市及在研药物盘点 简介 人类利用动物毒素治疗某些疾病有着悠久的历史
原标题:从敌人到朋友,动物毒素类上市及在研药物盘点

简介
人类利用动物毒素治疗某些疾病有着悠久的历史。早在公元前第七世纪,阿育吠陀医学(Ayurvedic medicine)中,蛇毒已经被用来延长生命和治疗关节炎及胃肠道疾病。一千多年来,中医将蟾蜍皮肤分泌物(Chan Su)视为一种利尿剂、麻醉剂和抗癌药物。在墨西哥中部和南美洲原住民的传统医学中,捕鸟蛛(tarantulas)用于治疗诸如哮喘和癌症等疾病。自20世纪30年代以来,眼镜蛇蛇毒一直用于治疗哮喘、小儿麻痹症、多发性硬化症、风湿、重度疼痛和三叉神经痛。20世纪70年代,在巴西蝮蛇Bothrops jaracaca的毒液中发现药用活性多肽,在此基础上开发了“重磅炸弹”卡托普利(Captopril,降压药,ACE抑制剂),此后,基于毒素的药物发现才为人们所关注。
虽然卡托普利(Captopril)的发现使大家认识到动物毒液在新药研发中的价值,但过去几十年里,从动物毒液衍生的新分子实体(NMEs)一直相对稀少。有以下几点原因,首先,与微生物相比,很多有毒动物在实验室条件下很难获得和维持。其次,许多有毒动物(包括几乎所有的有毒无脊椎动物)体型较小,仅能提供少量的毒液,大大延缓了研究进度。第三,在上世纪八九十年代用于表征动物毒液成分的分析技术比较原始,而毒液的成分又很复杂,因此造成动物毒液的化学成分和药理作用的巨大多样性没有被充分的理解。幸运的是,随着技术进步,对动物毒液的高通量筛选以及结构和功能的表征大大促进了基于毒素的药物发现。
基于动物毒素的上市药物
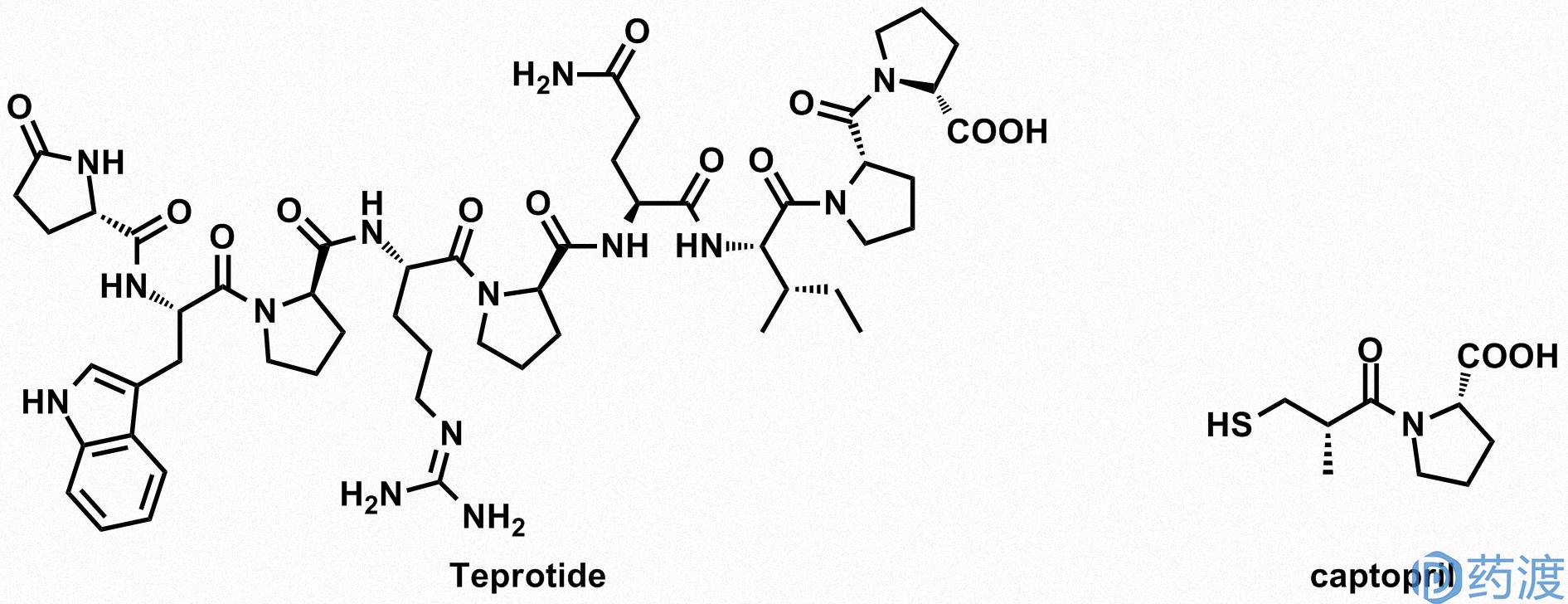
最早运用现代医疗理念和科技手段,将动物毒素开发成药物的实例是血管紧张素受体抑制剂(ACE抑制剂)卡托普利(Captopril)。1967年,牛津大学的John Vane和他的同事Mick Bakhle研究发现从巴西蝮蛇Bothrops jaracaca中提取的毒素在体外能有效的抑制血管紧张素受体。随后,John Vane和Squibb公司(现在是Bristol-Myers Squibb)合作对蛇毒提取液在心血管系统中的作用进行研究。Squibb公司的生物化学家David Cushman和有机化学家Miguel A. Ondetti从蛇毒中分离出了九肽化合物,替普罗肽(teprotide)。两人对替普罗肽(teprotide)的结构进行截取和改造,最终得到了卡托普利(Captopril),其对ACE的抑制活性增强了近2000倍。1981年,在此基础上,通过结构改造成功研发出可口服的卡托普利(Captopril),并成为治疗高血压的常规药物。卡托普利(Captopril)研发的成功极大鼓舞了医药企业从动物毒素中开发药物的热情和信心。
同一时期,国内也开发了一些酶类毒素药物,来源于蝰蛇和蛇岛蝮蛇的去纤酶等,也成功地应用于临床。蛇毒来源的酶类被用于血栓治疗,非酶类药物的研发进展很缓慢。直到1998年才研发出了作用于血小板上的糖蛋白Ⅱb/ Ⅲa整联蛋白受体的药物埃替非巴肽(Eptifibatide)和替罗非班(Tirofiban),它们可以抑制血小板聚集,从而防止血液凝固。在临床上,它们主要用于治疗心绞痛和急性心肌梗死。研究人员在蛇毒中发现了一种专一性糖蛋白Ⅱb/ Ⅲa整联蛋白受体抑制活性毒素,命名为Barbourin,该毒素序列中含有特定的RGD结构。随后根据Barbourin的结构,模拟设计出了环状七肽化合物埃替非巴肽(Eptifibatide)。而替罗非班(Tirofiban)则是根据从锯鳞蝰蛇(Echis carinatus)毒液中发现的锯鳞血抑肽(echistatin)为模板开发的,它能够抑制血纤维蛋白原依赖的血小板聚集。
近期成功开发的一些药物来源于具有丰富的结构和功能多样性的芋螺多肽毒素,例如2004年被FDA批准上市的齐考诺肽(Ziconotide),用于治疗多种慢性疼痛。该药特异性地作用于钙离子通道2.2亚型,可阻断沿脊髓传导的痛觉信号。
此外来源于水蛭唾液腺,用于治疗血栓的比伐卢定(Bivalirudin)和来源于蜥蜴唾液腺,用于治疗糖尿病的Exenatide也是成功案例。水蛭素(Hirudin)由65个氨基酸残基组成,是目前自然界中发现的活性最强凝血酶抑制剂。在解析水蛭素与凝血酶相互作用的结构基础和分子作用机制上,研究人员通过分子模拟和设计,开发了一类新的凝血酶抑制剂Hirulog。比伐卢定(Bivalirudin)是进一步根据水蛭素Hirulog的分子结构设计开发出的新型凝血酶抑制剂,它是由两个分别可以和凝血酶两个不同结合位点结合的肽段经过中间连接肽段连接而成的。目前,比伐卢定(Bivalirudin)是临床上使用最广泛的抗血栓药物之一。从大毒蜥蜴(Heloderma suspectum)的唾液中发现的胰高血糖素样肽-1受体(GLP-1)激动剂Exendin-4,是含有39个氨基酸残基的多肽。Exendin-4能与胰岛β细胞膜上的GLP-1受体结合,有很强的促胰岛素分泌作用。Exenatide就是人工合成的Exendin-4,2005年由FDA批准为治疗2型糖尿病药物。
20世纪80年代,昆明动物研究所的研究人员从中华眼镜蛇毒液中发现了一种蛇毒神经毒素(克痛宁,Cobratide)。研究发现,克痛宁(Cobratide)具有显著的镇痛活性,随后将其开发成高效的镇痛药物。在克痛宁注射液研制的基础上,开发了口服镇痛药药物“复方克痛宁(克洛曲片)”,其具有镇痛效果强、药效时间长、成瘾性小等特点。
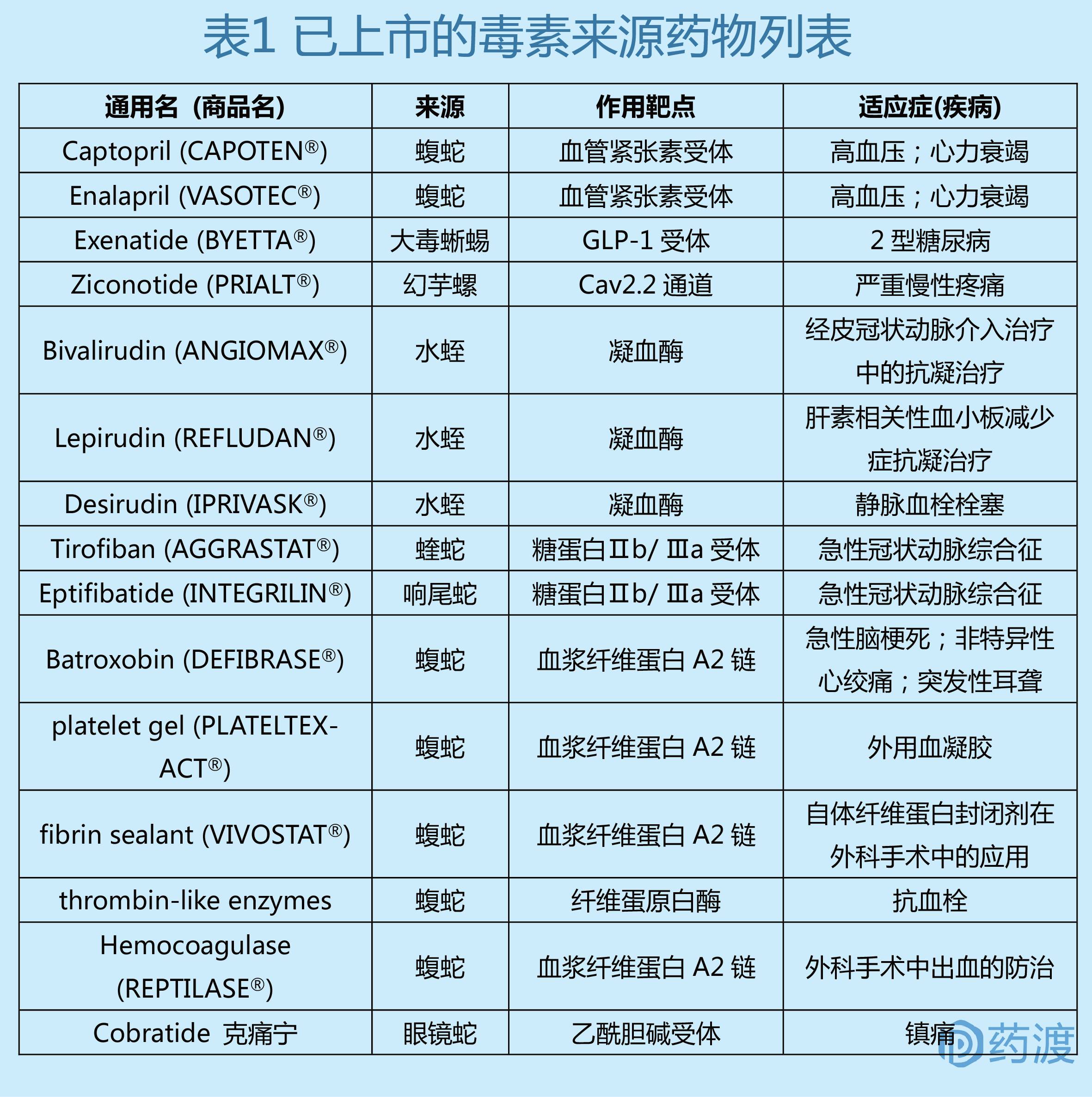
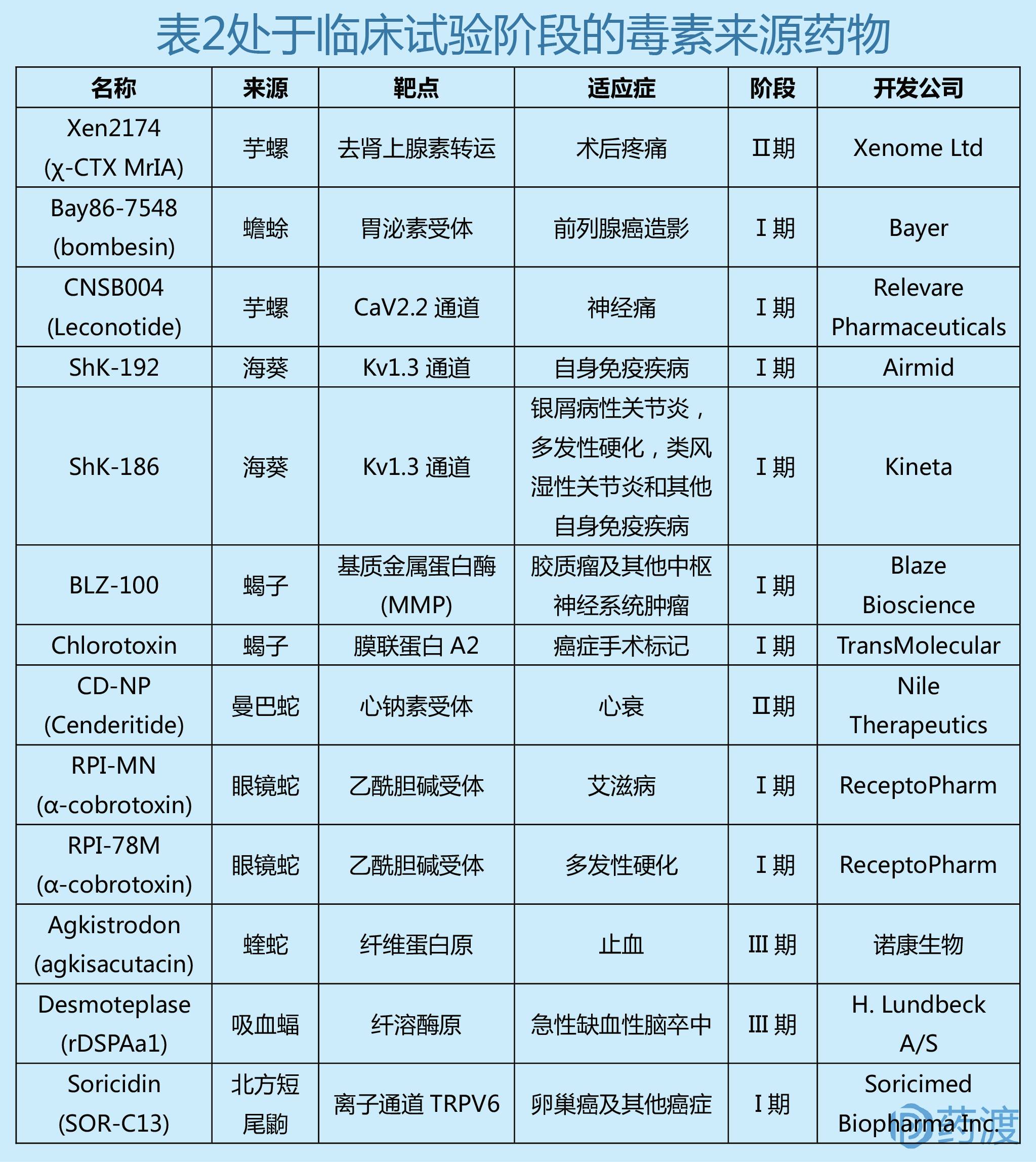
目前有15个临床上用于治疗多种疾病的毒液衍生药物,适应症涵盖高血压、疼痛和糖尿病,参见表1。
基于动物毒素的在研药物
现如今,有多种动物毒素来源的候选药物处于临床阶段(参见表2)。在发现齐考诺肽(Ziconotide)之后,研究人员还尝试对其他多种芋螺毒素进行开发,但因各种问题终止了临床研究。例如ω-conotoxin CVID的临床试验因为毒副作用而宣告失败;另一种芋螺毒素α-conotoxin Vc1.1,其作用机制较为复杂,起初被鉴定为乙酰胆碱受体抑制剂,之后的研究发现它可通过作用于GABA-B受体引起钙离子电流下降,从而抑制疼痛。然而,临床试验显示其疗效不佳。Xen2174是合成的芋螺毒素χ-conopeptide Mr1A的类似物,其可抑制神经元吸收肾上腺素,在动物实验中展示了很好的镇痛效果。虽然通过了Ⅰ期临床研究,但在之后的研究中发现,Xen2174具有剂量依赖性毒性,暂时情况不明。
从蛇毒中发现的心钠素类似物DNP与人源C-型心钠素结合成的新结构分子CD-NP的研究进展较为乐观。CD-NP可以激活心钠素受体A和B,其在慢性心衰病人体内的药动学、药效学、安全性、耐受性试验已完成,研究表明其在心肌梗死治疗中对左心室具有保护功能。海葵毒素来源的合成多肽ShK-186是一种钾离子通道抑制剂,正在进行早期临床试验。其可以通过抑制Kv1.3离子通道来治疗T-细胞自身免疫性疾病,如多发性硬化等。河豚毒素(Tetrodotoxin)可以抑制多种钠离子通道亚型,目前正在进行将其用于治疗晚期癌症疼痛的临床实验,现有数据结果显示河豚毒素能够明显减轻病人的疼痛反应。眼镜蛇来源的神经毒Cobratoxin经过结构修饰后可以刺激T-细胞的细胞因子分泌,目前正在进行将其用于治疗多发性硬化、运动神经元疾病、病毒感染的临床试验。
2000年,蒙特爱立森大学的Jack Stewart教授对北方短尾鼩(the northern short-tailed shrew)的唾液研究,发现了一种具有麻痹作用的多肽,命名为Soricidin。后续研究表明,Soricidin具有镇痛和抗肿瘤的活性,有可能被开发成非阿片类的镇痛药和抗癌药物。为此展开了两个研究项目,对Soricidin的结构进行表征和合成,并开发Soricidin的衍生物,以期能区分其抗肿瘤区域和镇痛区域。之后确定的抗肿瘤肽先导化合物对选择性钙离子通道TRPV6 (transient receptor potential vanilloid family number 6,TRPV6)有很好的抑制作用。这种离子通道在多种上皮细胞癌(如卵巢癌、前列腺癌、乳腺癌等)中过度表达,而在大多数健康组织中的表达水平较低。Soricidin衍生物(C系列肽)是该离子通道的高效抑制剂。在过度表达TRPV6的肿瘤细胞中,抑制TRPV6通道会中断特定的异常细胞信号传导途径,启动自我凋亡程序。在许多卵巢癌、前列腺癌和乳腺癌细胞系的体外研究中取得了非常成功的结果后,从较小的衍生化肽板中筛选出了候选药物。在确定没有明显毒性的情况下,在美国和加拿大开展了I期临床试验。
现在临床上使用的毒素来源药物大多是针对心血管系统疾病,而目前正在进行临床试验或临床前开发的毒素来源药物扩大了适应范围,包括慢性疼痛、自身免疫性疾病、伤口愈合、艾滋病和癌症,同时这些候选药物来自一个更广泛的有毒动物种类,包括蝙蝠、锥螺、海葵、蝎子、蛇和蜘蛛。
值得注意的是,目前已上市毒素来源药物的靶点多是酶,受体,或转运蛋白,只有齐考诺肽(Ziconotide)的靶点是离子通道。另一个引人注意的是对毒素蛋白和多肽二硫键结构的重视。目前在临床试验中的多数毒素衍生药物都含有1到14个二硫键。毒素蛋白通常注射到猎物的血液或软组织中,因此它们必须能够抵抗蛋白质水解。在某些情况下,需要穿过渗透障碍,如血脑屏障。毒素蛋白的一个普遍特征就是存在大量的半胱氨酸交联情况,这是因为半胱氨酸交联能提高分子稳定性和蛋白酶抗性。从成药性的角度来看,富含二硫键的毒素蛋白能使其在胃肠系统中存在更长时间,换句话说,更有可能开发成口服药物。
总结
虽然人类关注自然界的有毒动物和它们的毒液并进行探索已有数千年的历史,但真正从分子水平利用现代生物化学、生理学和生物物理学等技术对其进行研究不过半个多世纪。在短短五六十年的时间里,动物毒素的研究已取得了跨越式的发展,标志性的成果是发现了一批具有应用价值的动物毒素。这些发现不仅满足了人类的好奇心,也为人类的生活带来了可观的利益。现在我们仅触及了这个自然宝库的极小部分,相信未来一定会有更加令人兴奋的发现。
我国疆域辽阔,生态复杂,有着极其丰富的有毒动物资源。随着现代药物生物技术手段和分子生物学的发展以及对人类重大疾病机制认识的加深,我们极有可能在其中找到结构和功能全新的毒素,并在此基础上优化和设计出具有重要应用价值的临床药物,为广大的患者带来更多、更有疗效的药物。


- 好文

- 钦佩

- 喜欢

- 泪奔

- 可爱

- 思考













频道推荐

凤凰科技官方微信
视频
-

李咏珍贵私人照曝光:24岁结婚照甜蜜青涩
播放数:145391
-

金庸去世享年94岁,三版“小龙女”李若彤刘亦菲陈妍希悼念
播放数:3277
-

章泽天棒球写真旧照曝光 穿清华校服肤白貌美嫩出水
播放数:143449
-

老年痴呆男子走失10天 在离家1公里工地与工人同住
播放数:165128